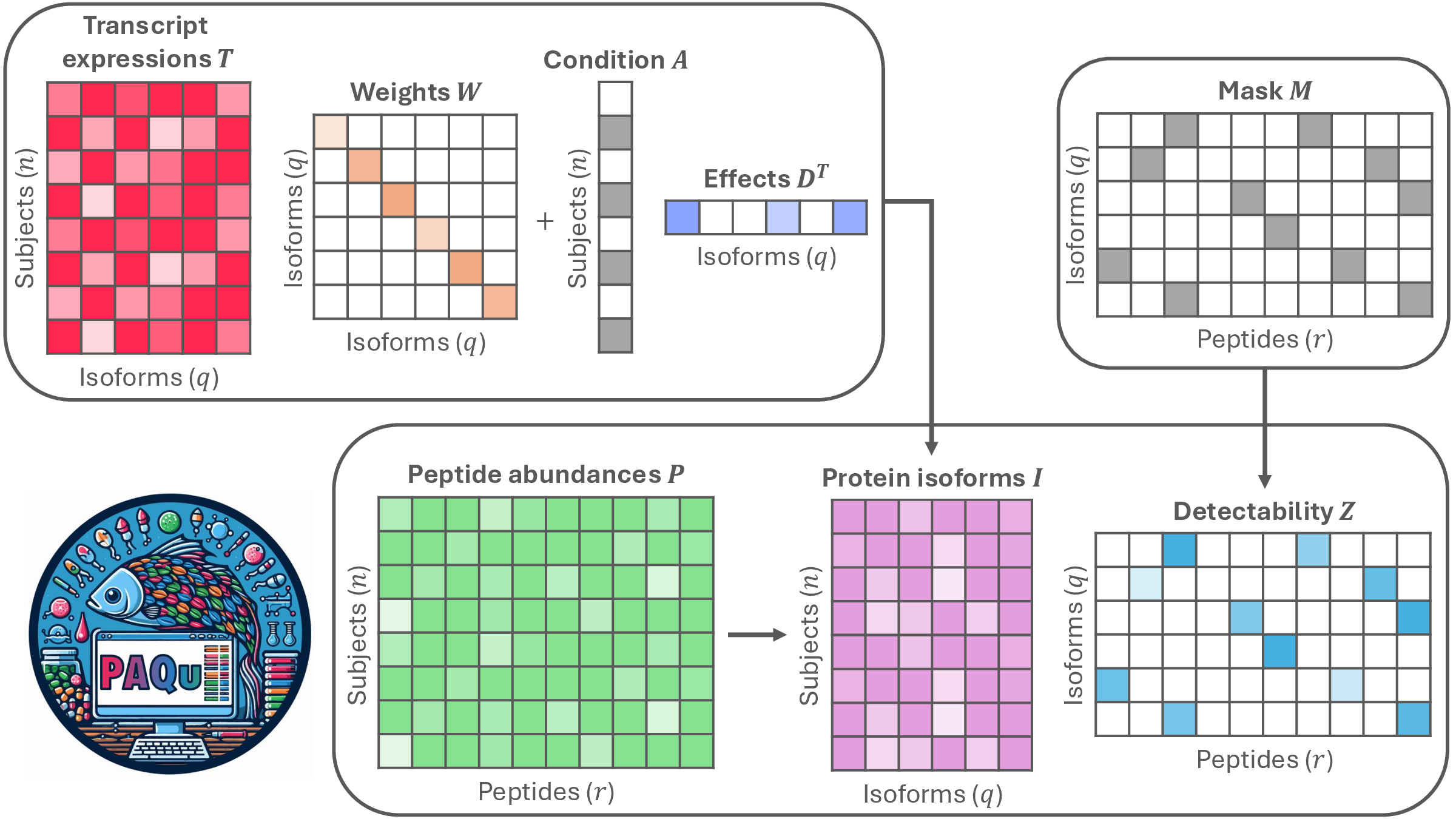
Estimating Protein Isoform Abundances with PAQu
A Bayesian supervised factor analysis method integrating transcriptomic and proteomic data for accurate, uncertainty-aware protein isoform quantification.
bioRxiv 2026
aDept. of Statistics & Data Science, Carnegie Mellon University · bL'EMbeDS, Sant'Anna School of Advanced Studies · cDept. of Psychiatry, University of Pittsburgh · dDept. of Statistics, University of Pittsburgh · eA2IDEA · fCenter for Neuroscience, University of Pittsburgh · gDept. of Computational Biology, Carnegie Mellon University · hBiomedical Mass Spectrometry Center, University of Pittsburgh